Jonathan Tennyson, Joanne Carr, M A Lysaght, Jimena Gorfinkiel, Zdeněk Mašínand Jakub Benda and many others. Debugging: Thomas Meltzer and others.
1996 - 2019
Introduction
CONGEN is a stand-alone program from the UKRmol+ package that is mostly used together with SCATCI. The purpose of CONGEN is to combine individual molecular orbitals calculated by quantum chemistry structure packages (and augmented with continuum orbitals in scatci_integrals), given their spatial symmetry properties, so that the resulting many-electron configurations have a given value of total spin and a particular spatial symmetry that is in accord with the point group of symmetries of the molecule being studied. The first step in the SCATCI algorithm [1] is based on the use of Yoshimine’s prototype configurations [2] it is only necessary to generate configuration spin functions (CSFs) corresponding to the first two continuum orbitals for each scattering symmetry.
In setting up a CONGEN calculation the overall symmetry of all CSFs (many-electron basis functions) must be specified. This is done in the first namelist, &state. A reference determinant is also given in this section relative to which all other determinants forming the CSFs will be stored.
The second namelist &wfngrp is used to select CSFs which are to be included in the N-particle basis. The procedure followed in the selection process is as follows:
- define a set of movable electrons,
- define the set of orbitals into which the movable electrons are to be distributed,
- place constraints on the CSFs generated.
Constraints can be placed upon which CSFs resulting from the distributions given will be accepted. Tests are made upon occupation relative to some reference occupancies. Constraints allow generation of very specific wave functions; nevertheless, in a typical run they are not used.
Several &wfngrp decks can be used. This allows the user to block the generated CSFs generated into groups.
Operation
Most of CONGEN routines are displayed in the following diagram, which indicates calls of one routine from another. The central subroutine is congen_driver::csfgen, and most of work is done in subroutines congen_distribution::distrb and congen_projection::projec (and code called therefrom). The light brown boxes in the diagram indicate routines that deal mostly with I/O, the white-colour routines are shared with other UKRmol+ packages, while the green-colour ones contain most of the algorithmic functionality of CONGEN.
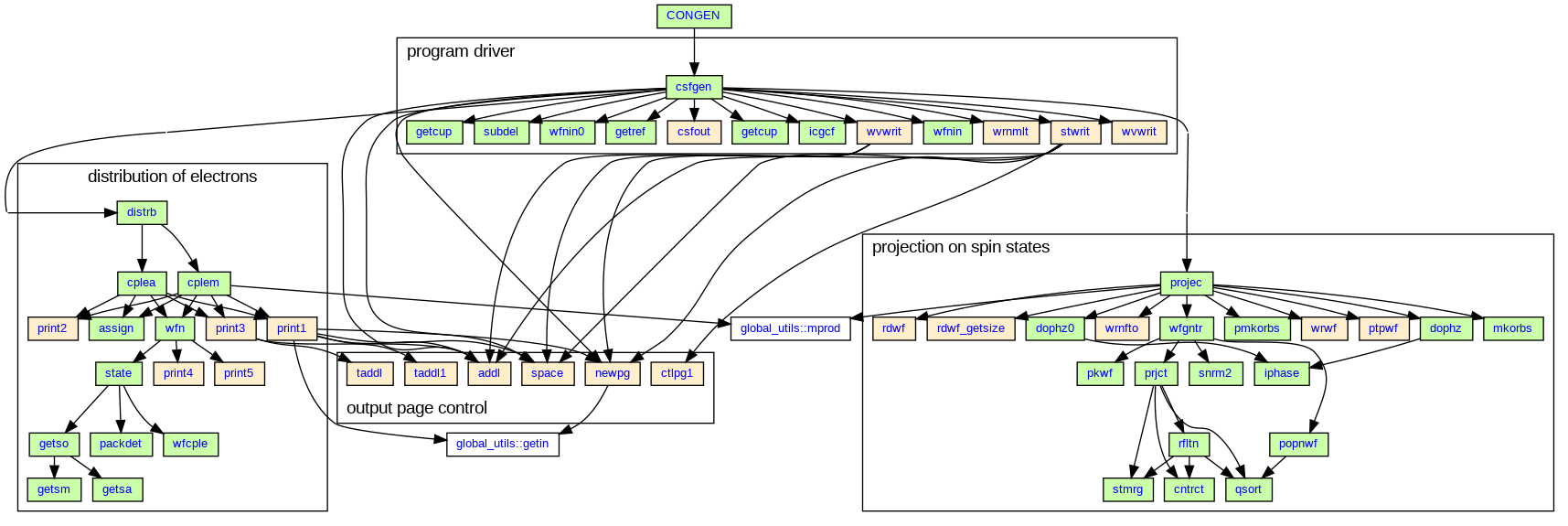
The program operates in the following way:
- Sensible initial (default) values are assigned to all variables. See also the next section.
- The input namelist
&stateis read. If it does not exist, program terminates. - The read data are checked for consistency, and some derived defaults are assumed.
- Maximal occupancy of shells of individual symmetries is set based on the point group according to this table:
Group v \ IRR > 1st 2nd 3rd .... C_infv 2 4 4 4 D_infh 2 2 4 4 other 2 2 2 2 - Reference determinant is formed (subroutine congen_driver::getref). This is an integer array that assigns a specific spin-orbital index to every electron. It corresponds to an anti-symmetrized non-degenerate linear combination of those spin-orbitals. The particular choice of spin-orbitals (what symmetry, what excitation, what spin, etc.) is totally in the hands of the user, via the namelist
&state. - Workspace (
mbx) is allocated on heap and partitioned into individual data arrays. - The so far constructed setup is printed, together with information about the reference determinant (subroutine congen_driver::stwrit).
- If requested (via
&state), the CSFs are read directly from the input stream (subroutine congen_driver::subdel). - The
&wfngrpnamelists are read one after another and immediately processed. - The main part of the "processing" is the distribution of electrons into prescribed orbitals and construction of wave functions of the correct spatial symmetry. The distribution into orbitals happens in congen_distribution::distrb.
- For every unique distribution of electrons into prescribed orbitals and for every unique coupling scheme (also constructed in congen_distribution::distrb) the function congen_distribution::cplem (or in case of Dinfh and Cinfv congen_distribution::cplea) is called.
- ...
- Wave functions are written to the output file (subroutine congen_driver::csfout). This also happens during the process (subroutine congen_distribution::wfn), whenever there is a shortage of workspace.
- A projection is now needed, so that the obtained determinants are eigenstates of the total spin. This happens in the subroutine congen_projection::wfgntr (called from congen_projection::projec). This is not necessary if there is no composition of spins (or it is trivial) – when all shells are closed or when open shells contain just one electron. However, when there are two or more electrons in open shells, all their spin permutations need to be added to the set of determinants (done in congen_projection::prjct).
- If requested, the phase correction for SCATCI is done, either in congen_projection::dophz0 (for target state calculation) or in congen_projection::dophz (for scattering state calculation).
- The header and projected wave functions are written back into the output file, replacing its previous contents.
Inputs
There are two namelist inputs. The first &state gives the namelist data which is common to all CSFs to be generated, i.e., all &wfngrp decks to follow. More than one &wfngrp can follow. Each &wfngrp generates a set of CSFs. The information in the previous &wfngrp is used as default for the following &wfngrp except for qntar, nelecp, gname, and defltc.
The variables that can be set in &state are in the following table, sorted in alphabetical order:
| variable | datatype | default value | description |
|---|---|---|---|
byproj | I | 1 | Project wave functions in 1 = CONGEN, 0 = SPEEDY, -1 = nowhere (should be avoided). |
cdimx | I | 400 | |
confpf | I | 1 | Print flag for the amount of print given of configurations and prototypes, see below. |
gutot | I | 0 | Inversion symmetry, used for D_infh only (+1 = gerade, -1 = ungerade). |
idiag | I | 0 (when iscat <= 0), or 1 (when iscat > 0) | Matrix element evaluation flag passed to SPEEDY or SCATCI, see below. |
lcdo | I | 500 | Limit on number of determinants. |
lcdt | I | 500 | Not used. |
lndo | I | 5000 | Limit on number of determinants. |
lndt | I | 5000 | Not used. |
iposit | I | 0 | Exotic particle flag: 0 = electrons only, +/-1 = positive non-electron, +/-2 = negative non-electron; + = separate, - = identical L^2 orbitals to those of electrons. |
lpp | I | 60 | Length of line printer page. Used to determine where to put page banner. |
iscat | I | 0 | Output format: 0 = SPEEDY, 1 = SCATCI, 2 = SCATCI + information for phase correcting target CI wavefunctions. |
isz | I | qntot(1) | 2*Sz + 1 |
ltri | I | 300 | |
megu | I | 14 | Output data set with the namelist input to be passed to SPEEDY. |
megul | I | 13 | File unit for binary output of generated configutrations (see section Outputs). |
nbmx | I | 2000000 | Workspace: Number of real numbers. |
ndimx | I | 4000 | |
nelect | I | 0 | Number of electrons in the system. |
nfto | I | 15 | Fortran data set number for output unit of SPEEDY. |
nftw | I | 6 (= stdout) | Standard output unit for program log redirection. |
nob | nI | 0 for all components | Number of molecular orbitals per irreducible representation (n = number of them) for use in configurations. |
nob0 | nI | 0 for all components | As nob, but excluding continuum orbitals. This must be less than or equal to nob. |
nobe | nI | 0 for all components (automatic) | Number of electronic orbitals. Together with nobp should give nob. |
nobp | nI | 0 for all components (automatic) | Number of positronic orbitals. Together with nobe should give nob. |
nobv | nI | 0 for all components (automatic) | Not used, currently just copy of nob0. |
nodimx | I | 100 | |
nndel | I | 0 | If greater than zero, read CSFs directly from input stream. |
npflg | 6I | 0 for all components | Print flags for SPEEDY / SCATCI, see below. |
npmult | I | 0 | Print flag for the D2h multiplication table (0 = no, 1 = yes). |
nrefo | I | 0 | How many quintets are needed to define the reference determinant (= value of "n" in sname below). |
nrerun | I | 0 | Restart flag for SPEEDY (not used in SCATCI). |
ntgsym | I | Maximun number of target state symmetries. Needed if iscat = 2 and qntar is used to constrain the coupling of L^2 CSFs. | |
qmoln | L | .false. | Execution of CONGEN from Quantenmol (provides some additional output). |
qntot | 3I | -2 for all components | Total quantum numbers: spin multiplicity (1 = single, 3 = triplet), spatial symmetry irreducible representation (starting from 0, up to 7 for D2h), mirror symmetry (+1 or -1). |
refgu | nI | -2 for all components | Gerade (= +1) or underage (= -1) symmetry for the M value of each reference quintet. Used only for D_infh molecules. |
reforb | n(5I) | -2 for all components | Reference determinant. All other determinants are internally stored as difference with respect to this one. Number of quintets (n) given by nrefo above. See below for detailed description. |
sname | A80 | blank string | Arbitrary text line that will be written to headers of all output pages. |
symtyp | I | -1 | Symmetry type, 0 = C_infv, 1 = D_infh, 2 = {D_2h, C_2v, C_s, E}. |
thres | WP | 1E-10 | Threshold for storing coefficients of matrix elements that contribute to Hij. |
The possible values of confpf are:
- 1: Minimum print; number of states for each prototype
- 10: PQN (see description of PQN in next namelist) for each configuration plus the above
- 20: PQN for each state plus the above
- 30: PQN for each state and determinant for each prototype plus the above
- 40: every configuration is given in terms of determinants plus the above
The possible values of idiag are:
- 0: Evaluate matrix elements as difference from first element. Do not use this option with SCATCI option
iexpc= 1. - 1: Evaluate all matrix elements in full.
- 2: Do not evaluate pure target integrals. And with automatic target generation, evaluate target matrix elements as difference from first element.
- 3: Do not evaluate pure target integrals. The value
idiag= 2 is strongly recommended with SCATCI optionicitg= 1, but must not be used with SPEEDY or for target only calculations. The entrynob0must be set for optionsidiag= 2 or 3. Note thatidiagcan be (re)set in using namelist&inputin SCATCI.
The possible values of npflg are:
npflg(1): 1 = print input CSF's in packed form; 0 = no print of CSF'snpflg(2): 1 = print open orbitals; 0 = no print of open orbitalsnpflg(3): 1 = print output CSF's in packed form; 0 = no print of output CSF'snpflg(4): 1 = print diagonal density expression, off diagonal density expression, and off diagonal energy expression; 0 = no print of these itemsnpflg(5): 1 = print target phases; 0 = no print / checking. For ISCAT = 0 usual calculationnpflg(6): 1 = print D2h multiplication table and integral pointers/boxes; 0 = do not print
The art of using CONGEN is in composition of the reference determinants, which has a very specific syntax. The parameter nrefo sets the number of quintets (packs of five integers) needed to contain the complete information. The array reforb then contains all these quintets joined together. Here is the explanation of the function of individual components of the quintets:
- 1st value: M-value (symmetry quantum number) of the group of orbitals to be described by this quintet.
- 2nd value: The serial number of first orbital of symmetry M to be filled for the group of orbitals described in this quintet.
- 3rd value: The number of electrons to be put into this shell filling each orbital to its maximum occupation if possible.
- 4th value: Zero for closed shell. For non-degenerate open shell: 0 = alpha, 1 = beta spin state. Otherwise see below.
- 5th value: Zero for both closed shell and for non-degenerate open shell. Otherwise see below.
For degenerate orbitals with two electrons or less the entries correspond to the occupied orbitals, the 4th value is assigned according to the following chart for the first electron in the open occupied orbital. The 5th value is assigned the corresponding value for the second electron. If the orbital is singly occupied, this last entry is set to zero.
= 0 lambda + alpha = 1 lambda + beta = 2 lambda - alpha = 3 lambda - beta
For more than two electrons in a degenerate orbital, the specification of the hole is given by the above rules. Examples:
- Pi_+alpha Pi_-alpha: (4) = 0, (5) = 2 - Theta_alpha: (4) = 0, (5) = 0 - Pi_+alpha Pi_+beta Pi_-beta: (4) = 2, (5) = 0
The variables that can be set in &wfngrp are in the following table, sorted in alphabetical order:
| variable | datatype | default value | description |
|---|---|---|---|
cup | User-specified intermediate couplings. See below for details. | ||
defltc | I | 0 | Use user-specified intermediate couplings between shells. |
gname | 80A | blank string | Arbitrary description of the setup. |
gushl | nshlp*I | -2 for all components | Used only for D_infh. Gives the gerage (= +1) and ungerade (= -1) character of the orbitals described in each pqn. |
lsquare | |||
mshl | nshlpI | For each pqn triplet give the symmetry quantum number of the set of orbitals described. The symmetry quantum numbers are the same as given in qntot. | |
nelecg | I | 0 | The total number of electrons which are movable. |
nelecp | ndprod*I | -1 for all components | The number of electrons which are to be distributed in each orbital set used for movable electrons. |
ndprod | I | 0 | The number of sets into which the orbitals, which are used for the distribution of movable electrons, are to be divided. |
npcupf | I | 0 | Print (= 1) or do not print (= 0) coupling information. |
nrcon | The number of replacements or occupancies that can deviate from the reference occupancies given in tcon for each constraint. | ||
nrefog | I | 0 | The number of quintets given in reforg needed to describe the movable electrons. |
nshcon | ntcon*I | Number of triplets required in tcon to describe the constraint. | |
nshlp | ndprod*I | 0 for all components | The number of pqn triplets needed to describe each set of orbitals. At least one triplet is given for each symmetry in each set. |
ntcon | I | 0 | Number of constraints to be given (max 10). |
pqn | nshlp*(3I) | Series of triplets needed to describe each set of orbitals. See below for details. | |
qntar | 3I | -1,0,0 | Controls the coupling of the first N-1 electrons as defined in the pqn groups to a state of total symmetry as specified by qntar. |
refgug | nrefog*I | -2 for all components | Used for D_infh to give the gerage (= +1) or ungerade (= -1) symmetry for the M values of each reference quintet. |
reforg | nrefog*(5I) | -2 for all components | Series of quintets which describe the movable electrons in the reference determinant. |
tcon | Constraints, see below. | ||
test | Test signals which of the two types of constraints will be applied. |
The integer array cup allows the user to specify intermediate couplings. First each shell is assigned a serial number in the order n which it was described in pqn. Suppose there were N shells, then the user would specify N-1 couplings. One such coupling would have the following form:
cup = N_i, N_j, N+1,
which means shell N_i is coupled to shell N_j to form a new intermediate shell N+1. The N+1 shell would then be coupled to another one of the N shells. The default coupling is simply that the first shell is coupled to the second to form an intermediate shell which is coupled to the third shell, etc.
The array pqn contains a series of triplets needed to describe each set of orbitals. At lease one triplet is needed for each symmetry in each set of orbitals. Each triplet will describe what will be referred to as a shell. The triplets can have two meanings which follow:
- When the 1st value of the triplet is zero then the other two represent the serial numbers of the initial and final orbitals in the set for that symmetry. (Serial numbers for orbitals are sequentially numbered starting with 1 in each symmetry at the 1st orbital of that symmetry.)
- When the 1st value of the triplet is non-zero, it is equal to the serial number of the orbital in the set. The second and third entries are then equal to zero.
Phase correction
One of partially hidden features of CONGEN is its ability to set up canonical "phases" of the configurations, which are later used by SCATCI (and MPI-SCATCI) to exactly match target CSFs generated in a target run of CONGEN to target CSFs generated in a scattering run of CONGEN. The evaluation of the phases is done in congen_projection::dophz0 and congen_projection::dophz for target and scattering systems, respectively. The canonical phase of a target CSF is the permutation sign of the first generated determinant in that CSF with respect to the reference determinant, multiplied by the sign of the coefficient of that determinant within the CSF. The formula is more complicated for the scattering run due to the additional continum orbitals; see the routines for detail.
These coefficients are also stored together with the resulting eigenvectors obtained from SCATCI (or MPI-SCATCI). Values of the phases will be displayed by CONGEN when setting npflg(5) = 1.
Outputs
CONGEN writes the calculated configurations to a single binary output file (in Fortran sequential unformatted format). The default is Fortran unit 13 (typically represented as the file fort.13), though this can be changed via the parameter megul. Several routines can write into that file, namely congen_driver::csfout, congen_distribution::wfn, congen_projection::wrnfto. The byte widths of the entries depend on how CONGEN was compiled. For instance, integers (denoted by I) can be either 4-byte or 8-byte. Similarly, the working precision real numbers (denoted by WP) can be either of single (4-byte), double (8-byte) or quadruple (16-byte) precision. This has to be kept in mind when sharing CONGEN, and more generally UKRmol+, outputs.
The output file structure is different when positrons are present in the configurations. The electron-only format is:
[A120] name, [I] mgvn, [I] S, [I] Sz, [WP] R,
[WP] pin, [I] norbw, [I] nsrbw, [I] nocsf, [I] nelt,
[I] lcdi, [I] idiag, [I] nsym, [I] symtyp, [I] lndi,
[6*I] npflg, [WP] thres, [I] nctarg, [I] ntgsym
IF ntgsym > 0:
[nctarg*I] iphz,
[nctarg*I] nctgt,
[ntgsym*I] notgt,
[ntgsym*I] mcont,
[ntgsym*I] gucont
IF ntgsym <= 0:
[nctarg*I] iphz
[nsym*I] nobw, [nelt*I] ndtrf, [nocsf*I] nodo, [I] iposit,
[nsym*I] nob0, [nx*I] nobl, [nx*I] nob0l,
[nsym*I] nobe, [nsym*I] nobp, [nsym*I] nobv
[m*I] icdo, [m*I] indo, [lndi*I] ndo, [lcdi*I] cdo
and many of these variables and dimensions are printed by CONGEN by default.
The electron + positron format is:
... TODO ...
References
[1] J. Tennyson, A new algorithm for Hamiltonian matrix construction in electron-molecule collision calculations, J. Phys. B: At. Mol. Opt. Phys. 29 (1996) 1817–1828.
[2] M. Yoshimine, Construction of Hamiltonian matrix in large configuration interaction calculations, J. Comput. Phys. 11 (1973) 449–454.